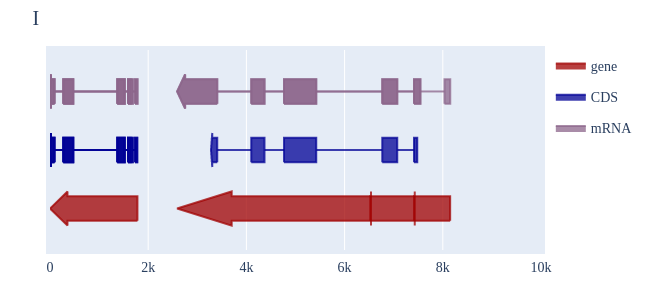
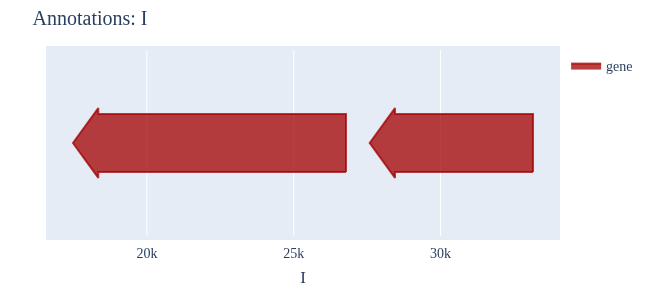
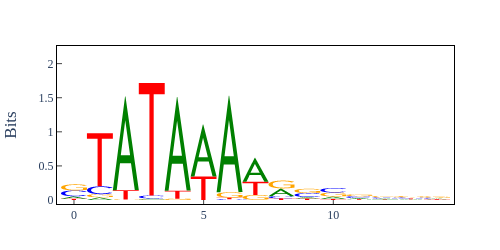
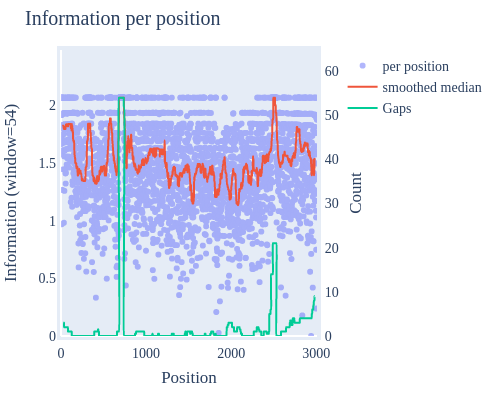
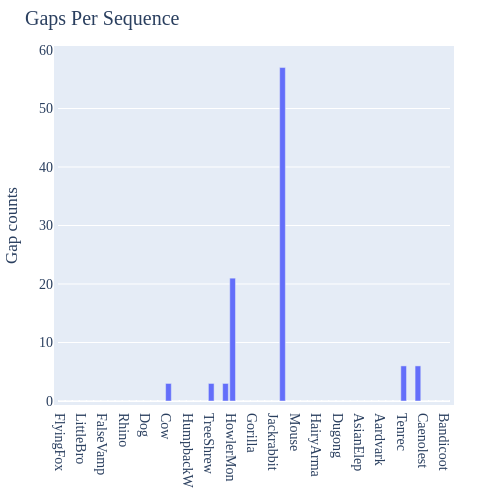
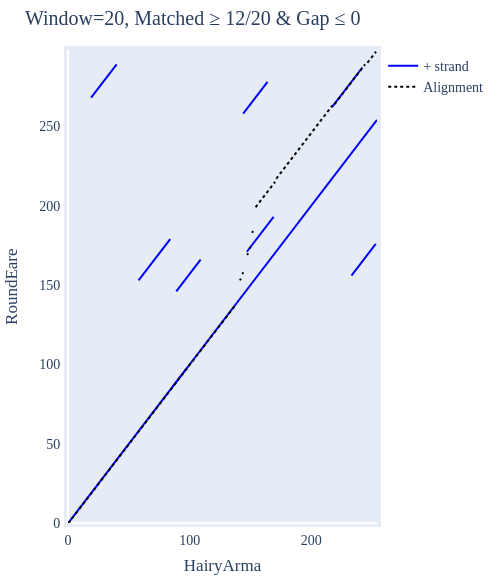
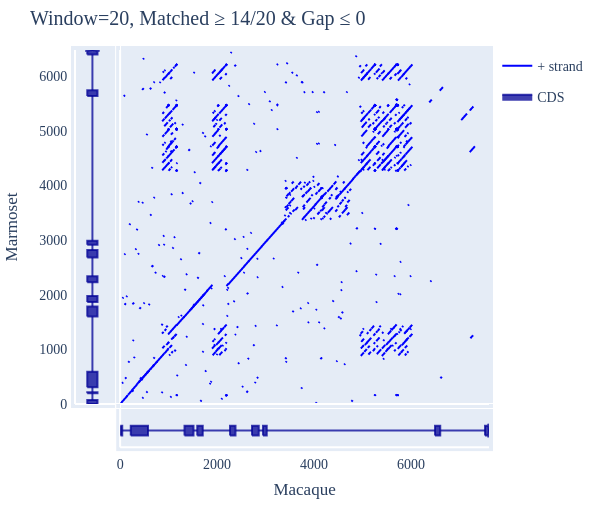
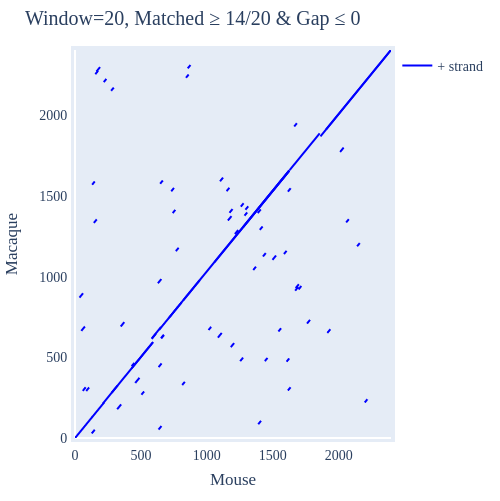
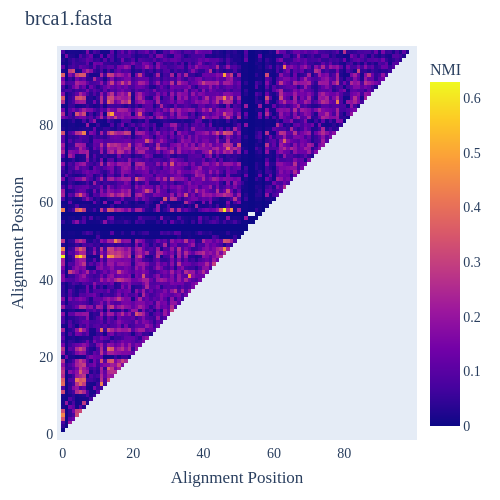
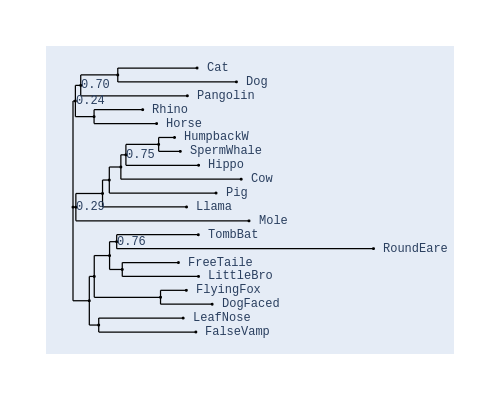
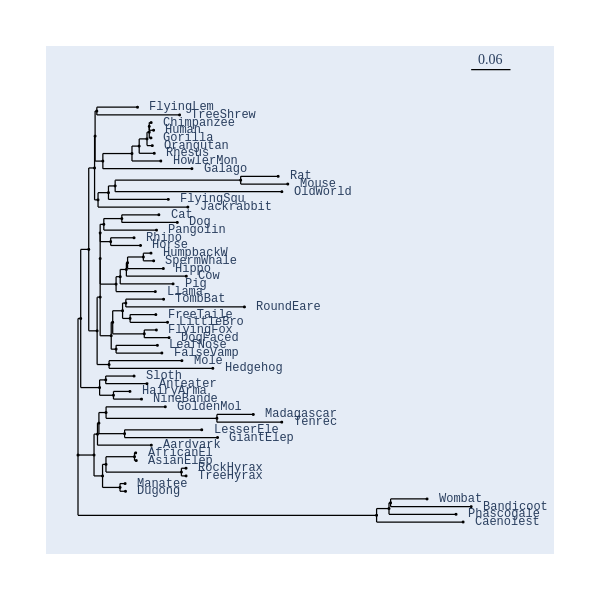
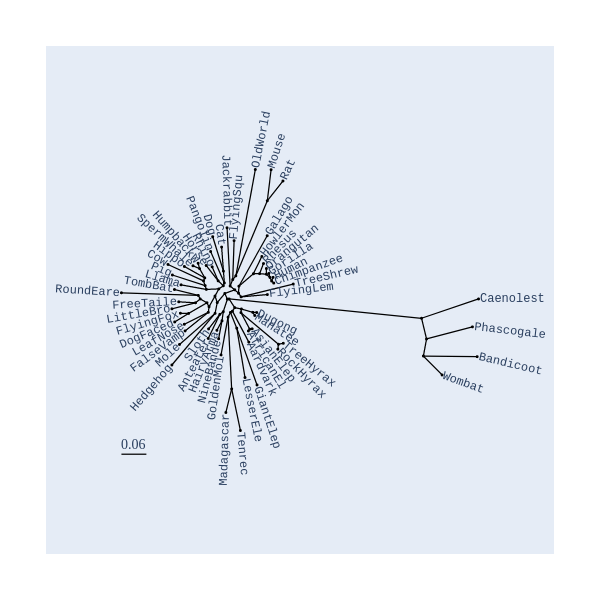
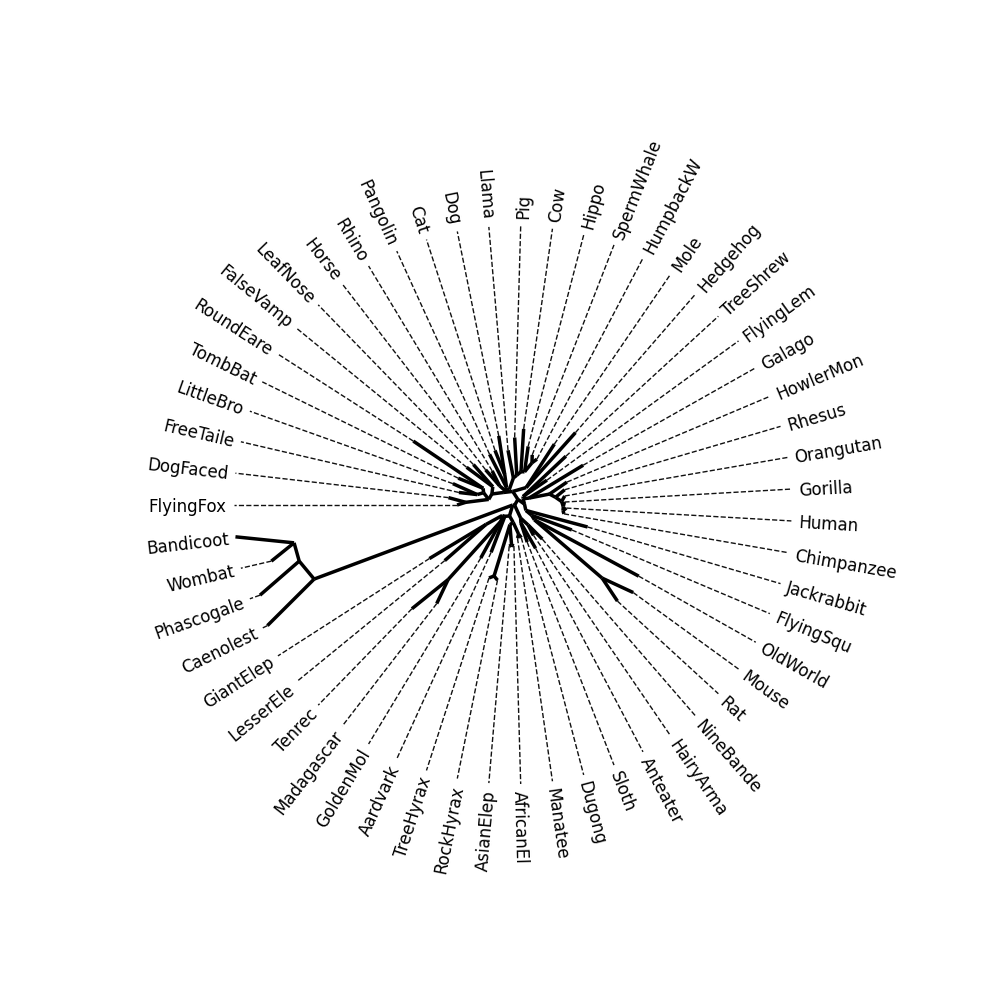
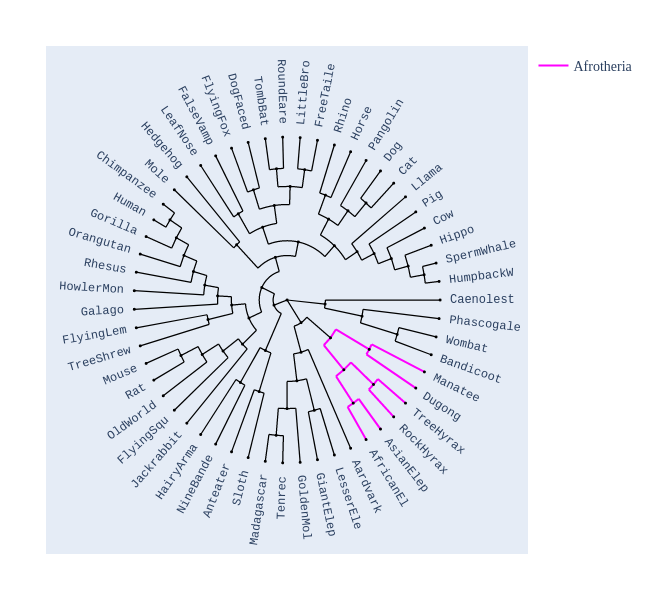
Image Gallery# We use Plotly as our backend for visualisation. It provides excellent graph interactivity in Jupyter notebooks. Annotations# Sequence Features Annotation DB Features Alignments & Sequences# Sequence logos Information analysis of an alignment Counting gaps per sequence Assess alignment quality via dotplots Dotplot with annotated sequences Dotplot basics Coevolution analysis Phylogenetic Trees# Showing Bootstrap Support Square Dendrogram Style Radial Dendrogram Style Using iplotx with cogent3 Circular Dendrogram Style Angular Dendrogram Style